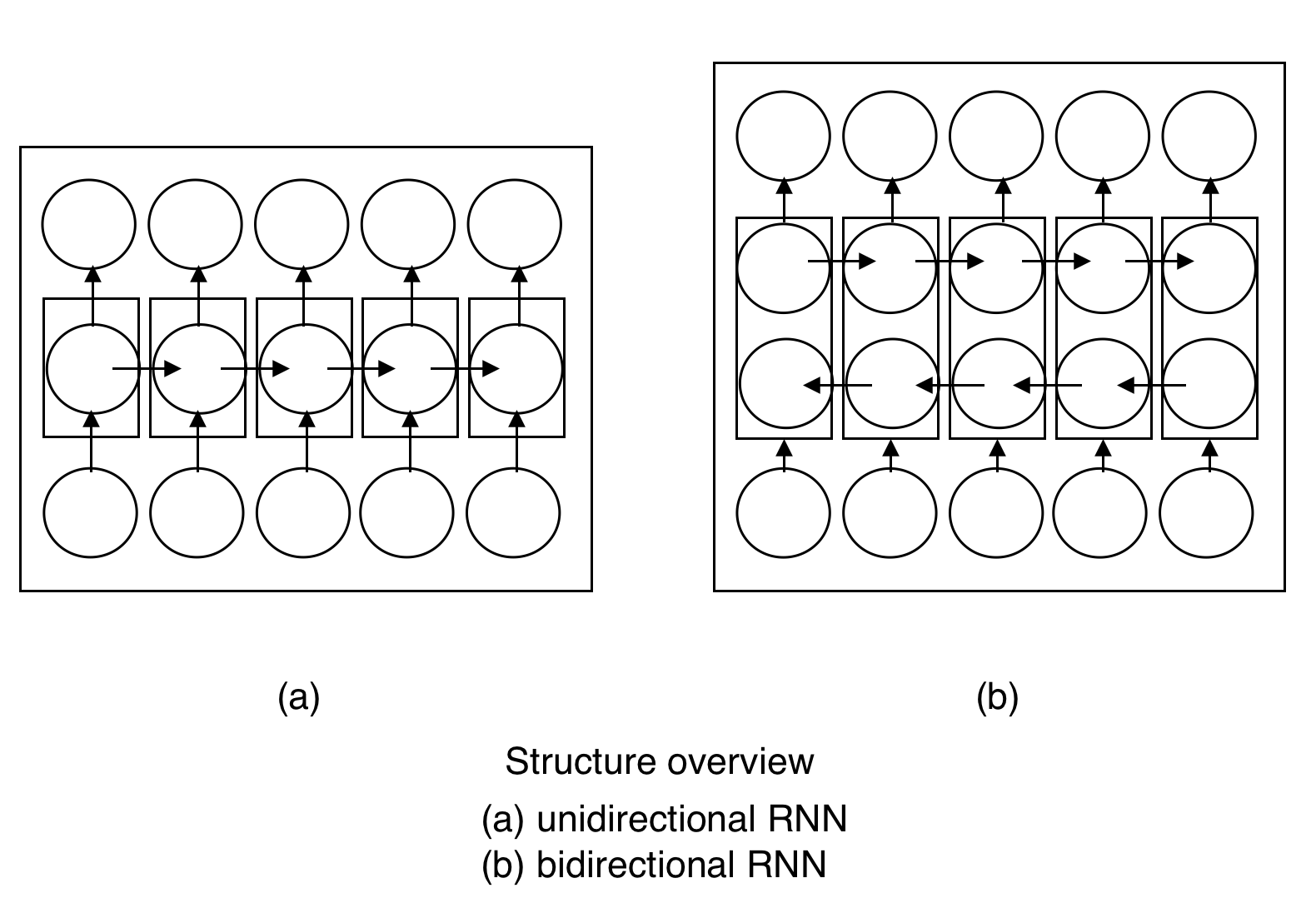
The predictor was trained using Bidirectional LSTM (Long Short-Term Memory), which is a kind of Recurrent Neural Network. It is useful for sequential data such that outputs of nearby amino acids in a sequence will have an influence on each other.
The dataset contains more than 394k amino acid sequences and their corresponding secondary structures.
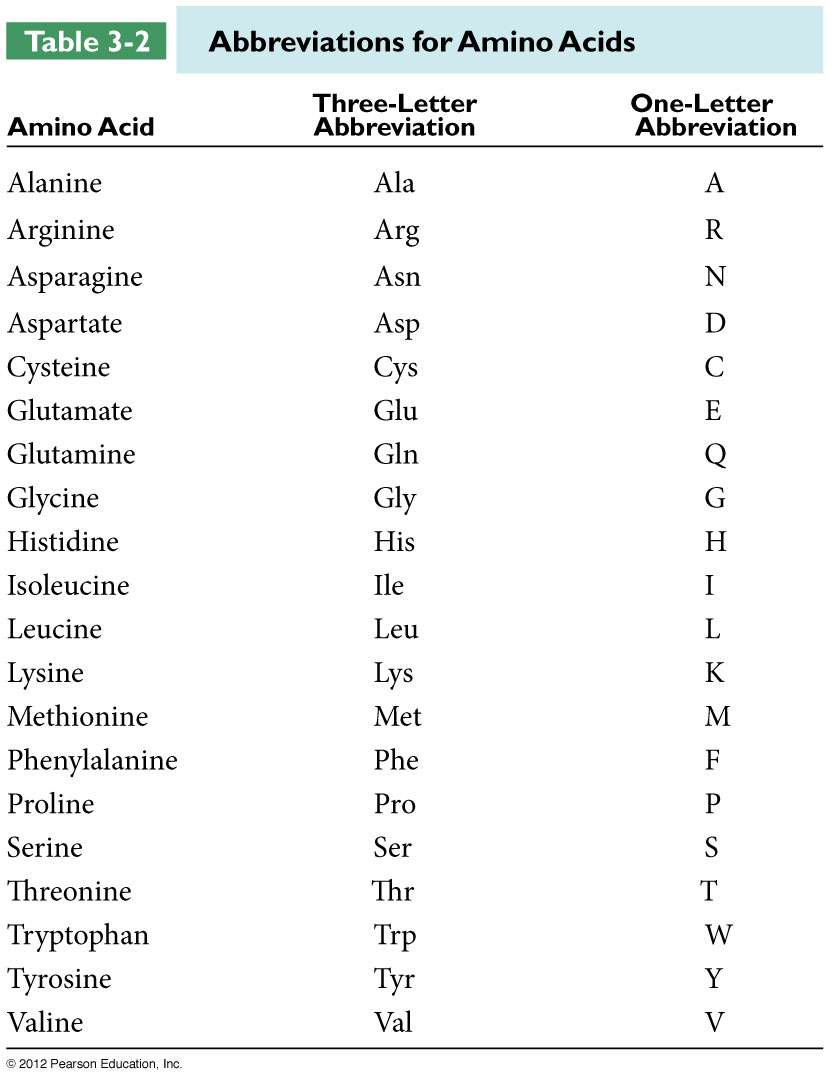
Amino acid sequence should be entered according to the one-letter codes.
The output will be their secondary structure represented by eight letters using DSSP (Define Secondary Structure of Proteins).
G = 3-turn helix. Min length 3 residues
H = 4-turn helix (α helix). Minimum length 4 residues
I = 5-turn helix (ϖ helix). Minimum length 5 residues
T = hydrogen bonded turn (3, 4, or 5 turns)
E = extended strand in parallel and/or anti-parallel β-sheet conformation. Min length 2 residues
B = residue in isolated β-bridge (single pair β-sheet hydrogen bond formation)
S = bend (the only non-hydrogen-bond based assignment)
C = coil (residues which are not in any of the above conformations)
We merge (E, B) into E, (H, G, I) into H, and (C, S, T) into C so that the ouput will only contain these three letters to represent more generalized structures.
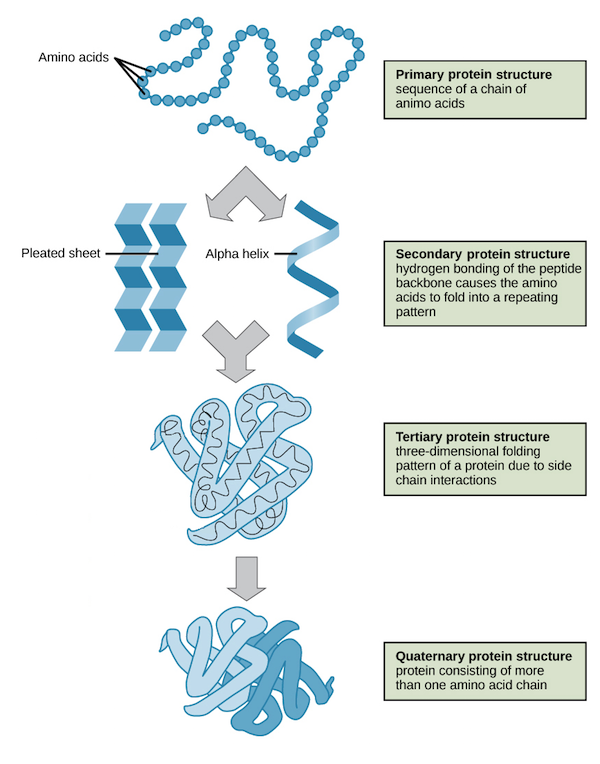
Proteins have four levels of structure: primary, secondary, tertiary, and quaternary.
This program predicts only the secondary structure what refers to local folded structures that form within a polypeptide due to interactions between atoms of the backbone.